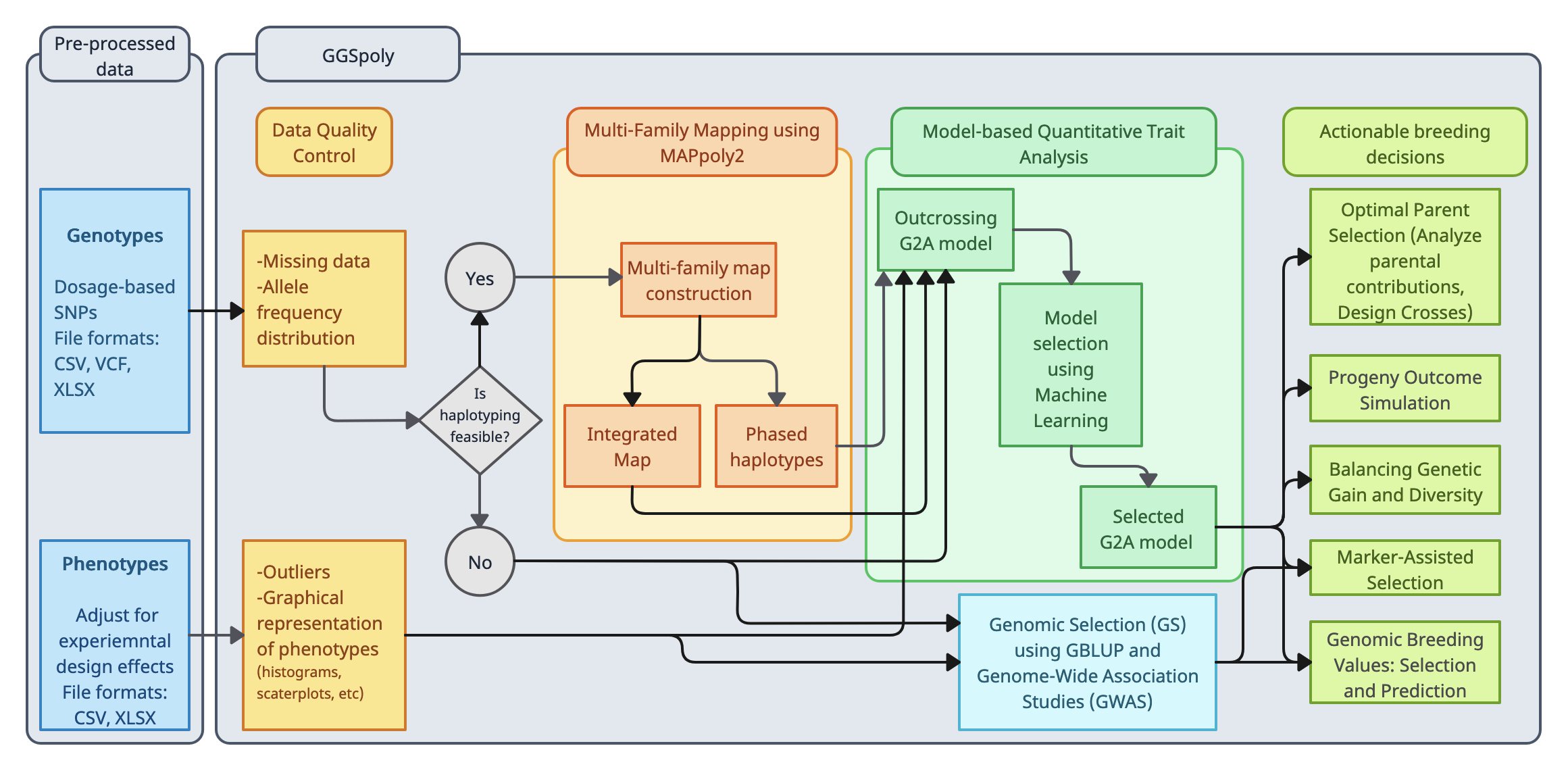
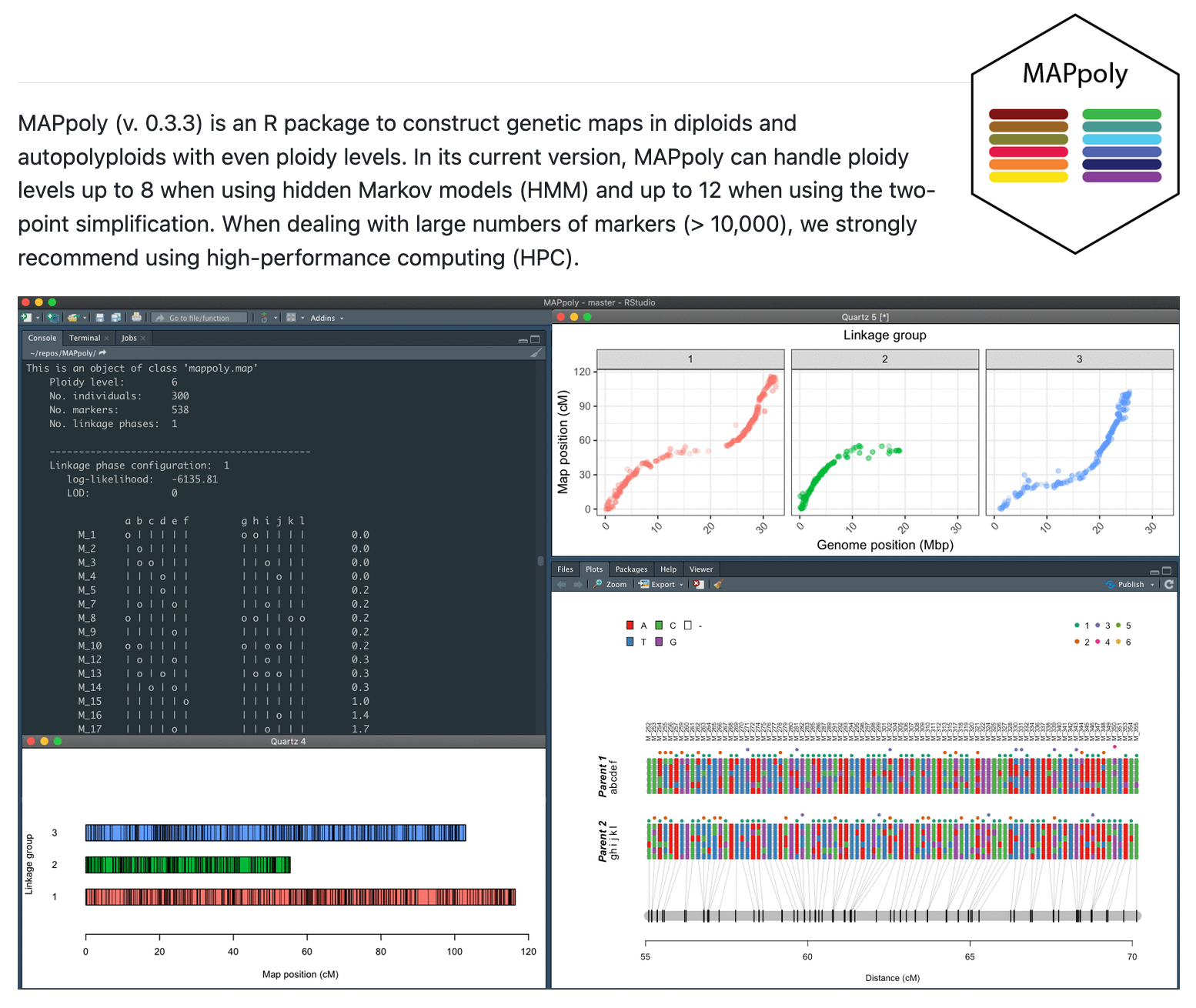
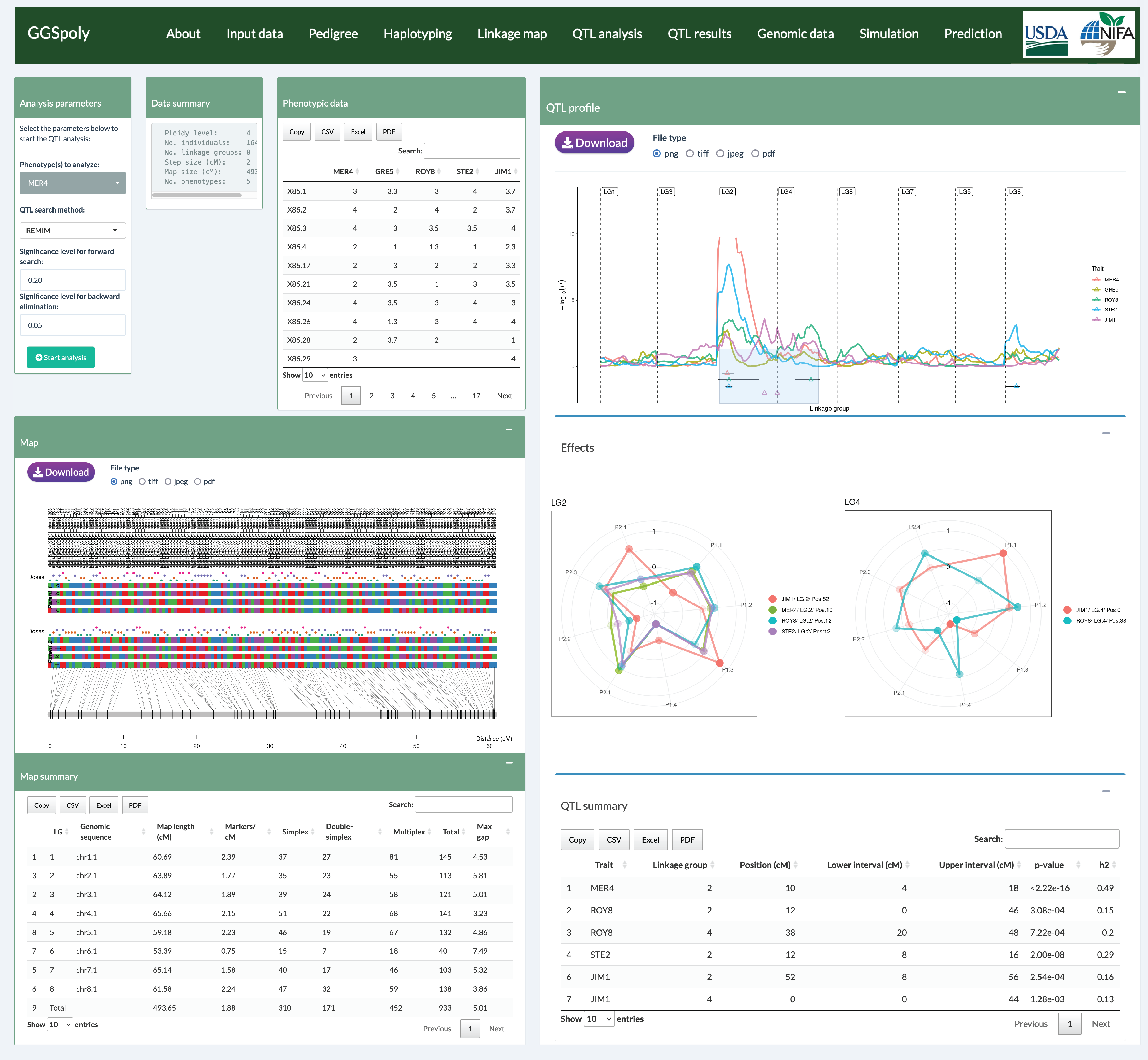
MAPpoly2 is an advanced R package designed for constructing genetic maps in
interconnected
full-sib autopolyploid families. This enhanced version prioritizes user-friendliness and
accessibility, with
improvements geared toward seamless integration with tools like R Shiny to
provide an intuitive
interface for polyploid genetic mapping. The package supports ploidy levels of 2, 4, and 6,
allowing for any
combination of these configurations, making it highly versatile for diverse research needs.
A standout feature of MAPpoly2 is its significantly enhanced performance,
achieved through
the integration of computationally intensive code implemented in C++. This
optimization enables
the efficient analysis of large and complex datasets, addressing a common bottleneck in
genetic mapping workflows.
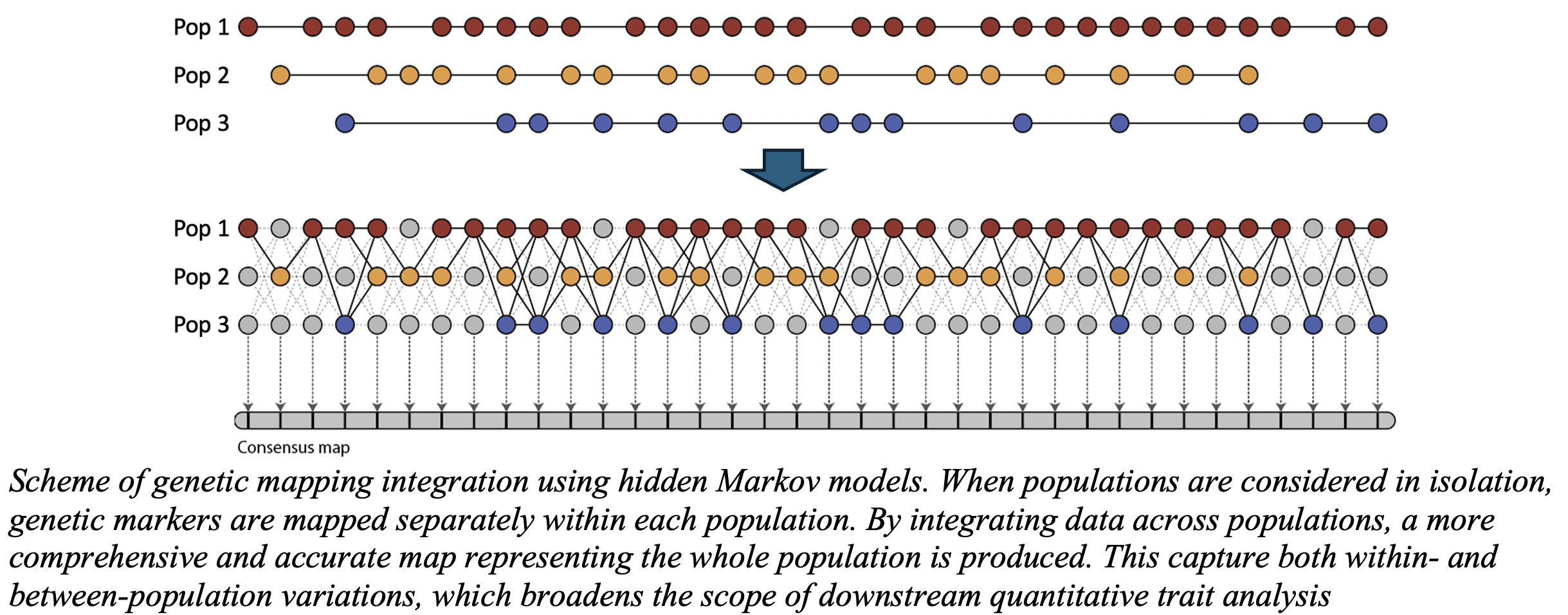
The package employs a robust Hidden Markov Model (HMM) framework to
construct individual
genetic maps for each parent. These maps are then combined into a comprehensive joint map,
which is recomputed
to incorporate any remaining markers, ensuring high accuracy and resolution in the final
results. These innovations
make MAPpoly2 an invaluable tool for researchers working with polyploid
species and large-scale
genomic datasets.